Higher-Order Equivariant Neural Networks for Charge Density Prediction in Materials
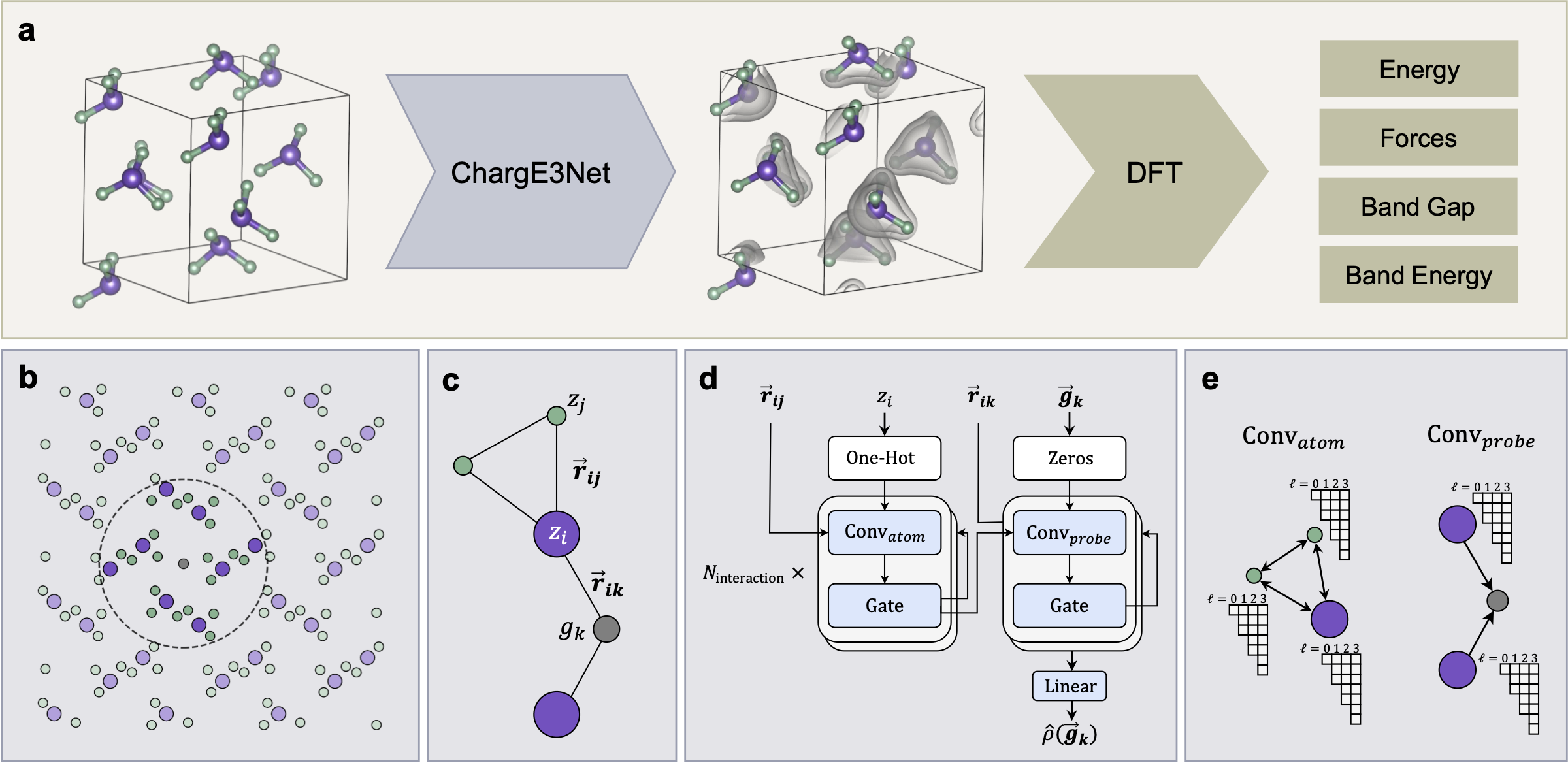
The calculation of electron density distribution using density functional theory (DFT) in materials and molecules is central to the study of their quantum and macro-scale properties, yet accurate and efficient calculation remains a long-standing challenge. We introduce ChargE3Net, an E(3)-equivariant graph neural network for predicting electron density in atomic systems. ChargE3Net enables the learning of higher-order equivariant feature to achieve high predictive accuracy and model expressivity. We show that ChargE3Net exceeds the performance of prior work on diverse sets of molecules and materials. When trained on the massive dataset of over 100K materials in the Materials Project database, our model is able to capture the complexity and variability in the data, leading to a significant 26.7% reduction in self-consistent iterations when used to initialize DFT calculations on unseen materials. Furthermore, we show that non-self-consistent DFT calculations using our predicted charge densities yield near-DFT performance on electronic and thermodynamic property prediction at a fraction of the computational cost. Further analysis attributes the greater predictive accuracy to improved modeling of systems with high angular variations. These results illuminate a pathway towards a machine learning-accelerated ab initio calculations for materials discovery.
PDF Abstract

 QM9
QM9
